Resource
| Id | pipeline/hg38_clinical_annotation |
|---|---|
| Type | annotation_pipeline |
| Version | 0 |
| Summary |
Clinical Annotation Pipeline for hg38
|
| Description |
This is a pipeline to annotate variants in hg38 assembly with Clinical resources. |
| Labels |
Pipeline Documentation
preamble
| Summary | Clinical Annotation Pipeline for hg38 |
|---|---|
| Description | This pipeline annotates hg38 variants with clinical resources. |
| Input reference genome | hg38/genomes/ucsc-hg38 |
Annotators
Worst effect across all transcripts.
Effect details for each affected transcript. Format: < transcript 1 >:<gene 1>:<effect 1>:<details 1>|...
<gene_1>:<effect_1>|... A gene can be repeated.
Annotator to identify the effect of the variant on protein coding.
Normalized allele.
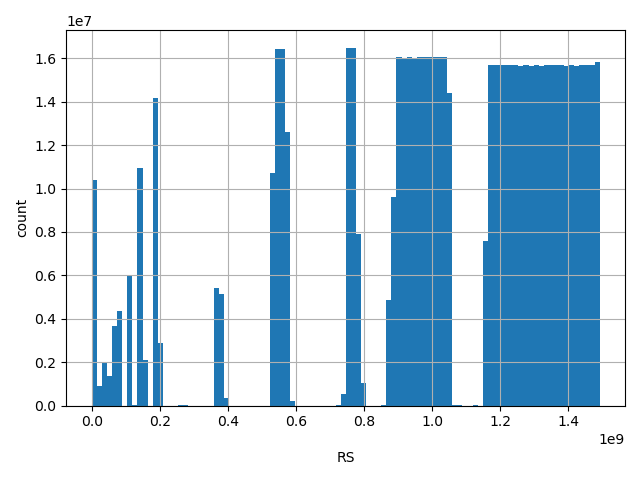
dbSNP ID (i.e. rs number)
allele_aggregator: list
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
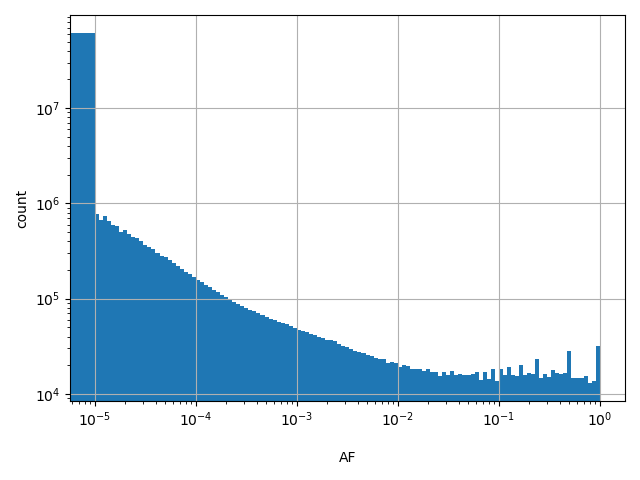
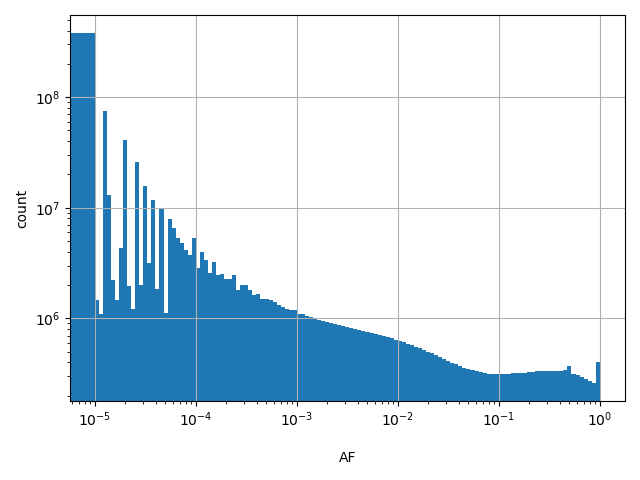
Alternate allele frequency
allele_aggregator: max
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
Alternate allele frequency
allele_aggregator: max
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
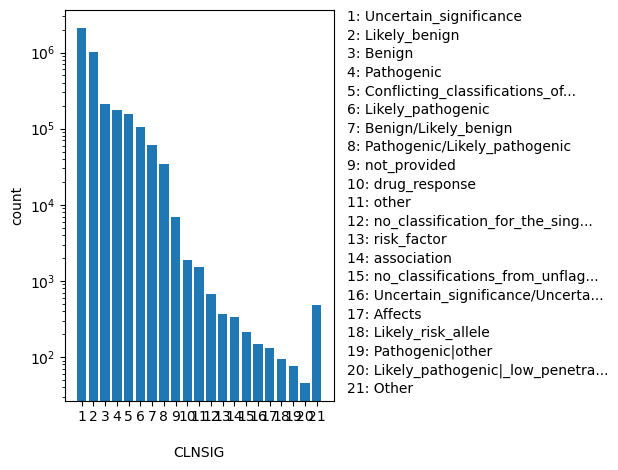
Aggregate germline classification for this single variant; multiple values are separated by a vertical bar
allele_aggregator: list
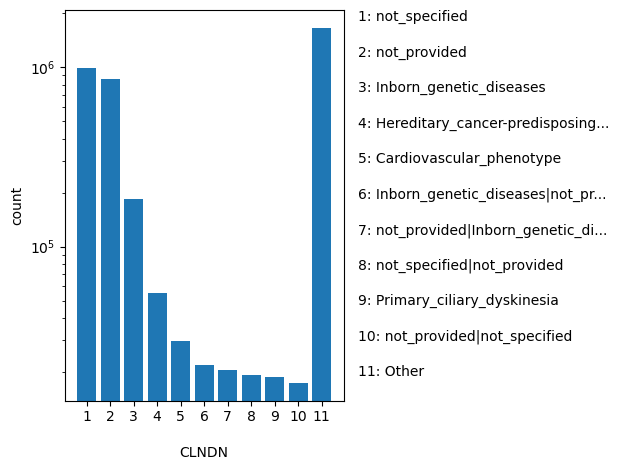
ClinVar's preferred disease name for the concept specified by disease identifiers in CLNDISDB
allele_aggregator: list
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
The score is a number that reflects the conservation at a position.
position_aggregator: mean [default]
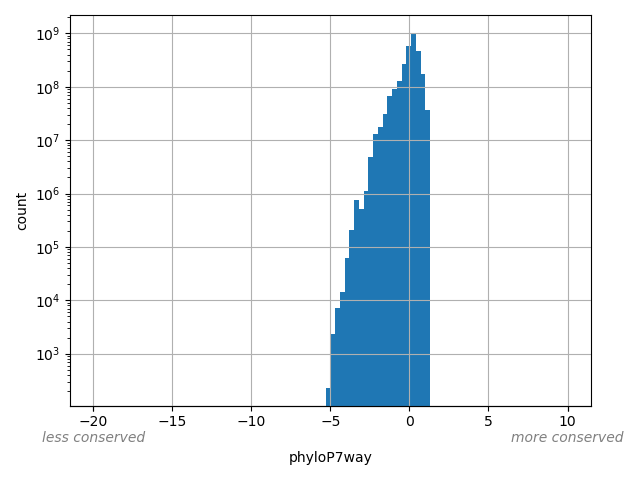
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
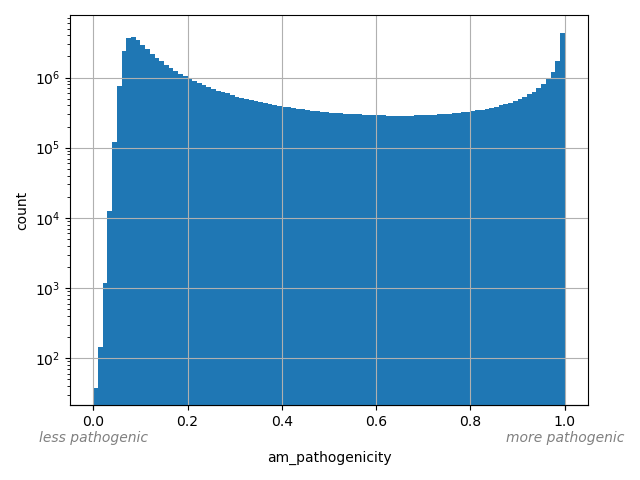
AlphaMissense Pathogenicity score is a deleteriousness score for missense variants
allele_aggregator: max
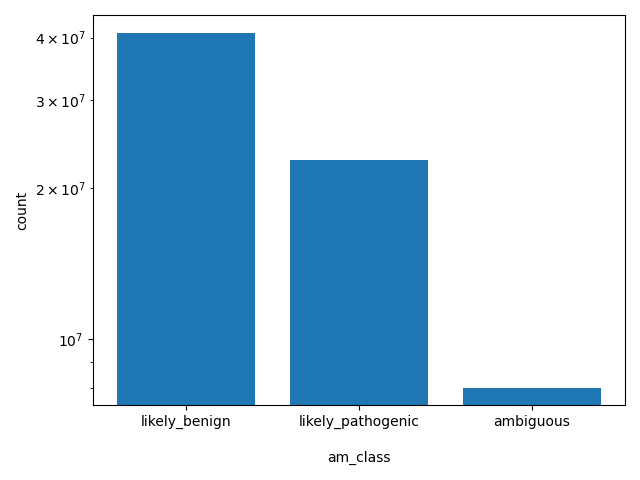
AlphaMissense Class is a deleteriousness category for missense variants
allele_aggregator: list
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
The lifted over annotatable
Annotator to lift over a variant from one reference genome to another.
Missense badness, PolyPhen-2, and Constraint. A deleteriousness prediction score for missense variants"
allele_aggregator: max
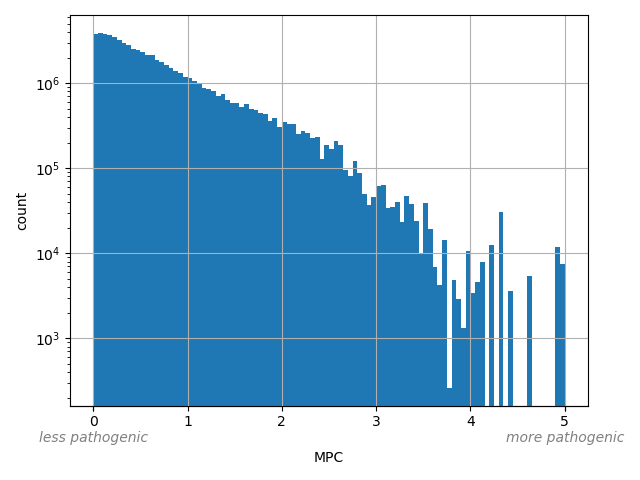
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
hg19_annotatable
Worst effect across all transcripts.
Effect details for each affected transcript. Format: < transcript 1 >:<gene 1>:<effect 1>:<details 1>|...
<gene_1>:<effect_1>|... A gene can be repeated.
List of all genes
Annotator to identify the effect of the variant on protein coding.
Gene rank after sorting by pLI intolerance score
Gene rank after sorting by pLI intolerance score
Gene ranks after sorting by LOEUF scores
Gene ranks after sorting by LOEUF scores
Files
| Filename | Size | md5 |
|---|---|---|
| genomic_resource.yaml | 237.0 B | c70460c66c53bdb446abcb4aad4f8404 |
| hg38_clinical_annotation.yaml | 2.82 KB | 00fd293d4213fbbef44fa1f18ca00f84 |
| statistics/ |