Resource
| Id | pipeline/T2T_clinical_annotation |
|---|---|
| Type | annotation_pipeline |
| Version | 0 |
| Summary |
Clinical Annotation Pipeline for T2T coordinates
|
| Description |
This is a pipeline to annotate variants from t2t assembly with Clinical resources. |
| Labels |
Pipeline Documentation
preamble
| Summary | Annotates t2t coordinates with Clinical resources |
|---|---|
| Description | This is a pipeline to annotate variants from t2t assembly with Clinical resources |
| Input reference genome | t2t/genomes/t2t-chm13v2.0 |
Annotators
The lifted over annotatable
Annotator to lift over a variant from one reference genome to another.
Normalized allele.
- input_annotatable:
hg38_annotatable
dbSNP ID (i.e. rs number)
allele_aggregator: list [default]
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
Alternate allele frequency
allele_aggregator: max [default]
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
Alternate allele frequency
allele_aggregator: max [default]
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
ClinVar's preferred disease name for the concept specified by disease identifiers in CLNDISDB
allele_aggregator: list [default]
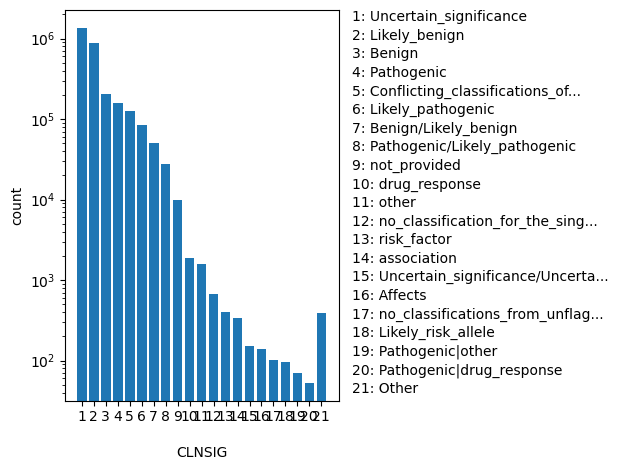
Aggregate germline classification for this single variant; multiple values are separated by a vertical bar
allele_aggregator: list [default]
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
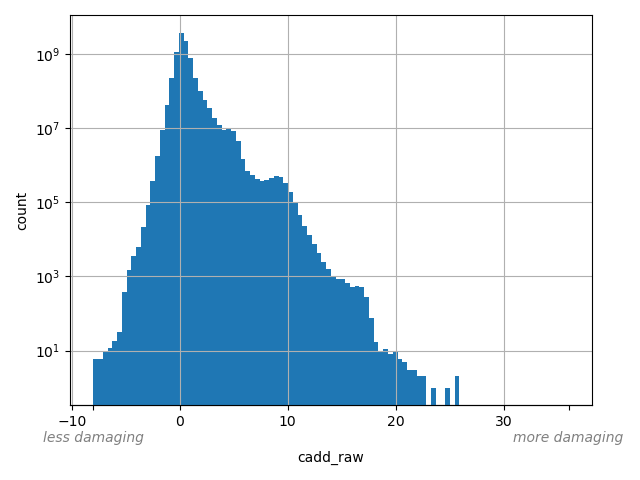
CADD raw score for functional prediction of a SNP. The larger the score the more likely the SNP has damaging effect
allele_aggregator: max [default]
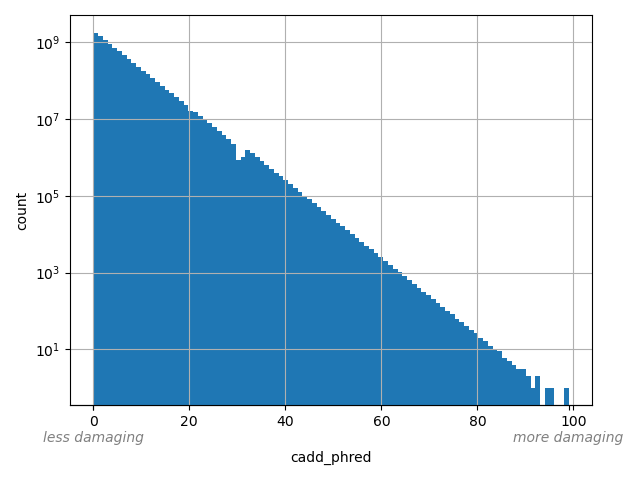
CADD phred-like score. This is phred-like rank score based on whole genome CADD raw scores. The larger the score the more likely the SNP has damaging effect.
allele_aggregator: max [default]
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
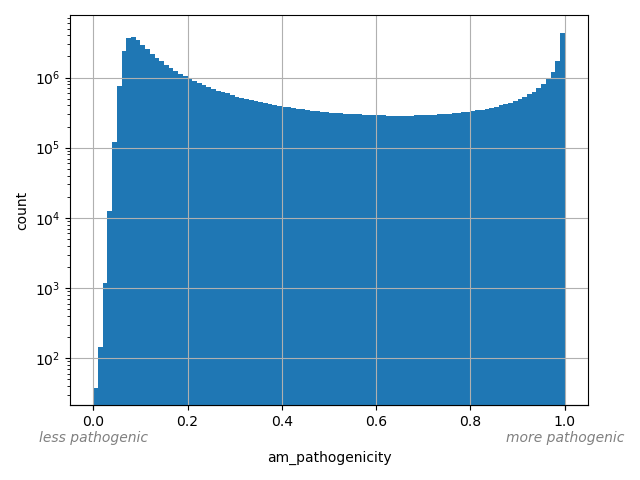
AlphaMissense Pathogenicity score is a deleteriousness score for missense variants
allele_aggregator: max [default]
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
The lifted over annotatable
Annotator to lift over a variant from one reference genome to another.
Missense badness, PolyPhen-2, and Constraint. A deleteriousness prediction score for missense variants"
allele_aggregator: max [default]
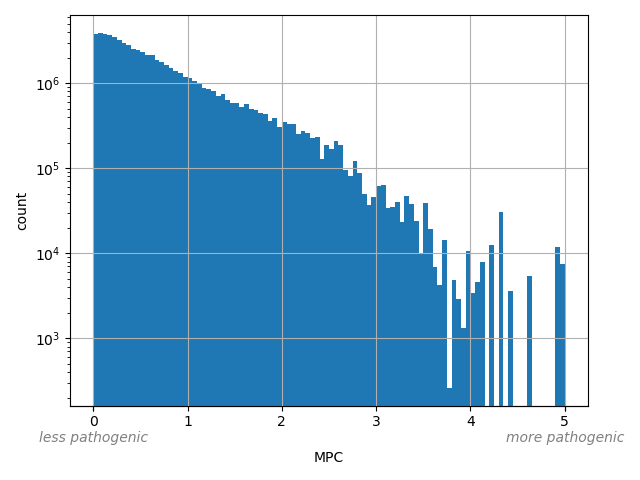
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
hg19_annotatable
Worst effect across all transcripts.
Effect details for each affected transcript. Format: < transcript 1 >:<gene 1>:<effect 1>:<details 1>|...
<gene_1>:<effect_1>|... A gene can be repeated.
List of all genes
Annotator to identify the effect of the variant on protein coding.
- input_annotatable:
normalized_allele
Worst effect across all transcripts.
Effect details for each affected transcript. Format: < transcript 1 >:<gene 1>:<effect 1>:<details 1>|...
<gene_1>:<effect_1>|... A gene can be repeated.
Annotator to identify the effect of the variant on protein coding.
- input_annotatable:
normalized_allele
Gene rank after sorting by pLI intolerance score
gene_aggregator: dict [default]
Gene rank after sorting by pLI intolerance score
gene_aggregator: min
Gene ranks after sorting by LOEUF scores
gene_aggregator: dict [default]
Gene ranks after sorting by LOEUF scores
gene_aggregator: min
Worst effect across all transcripts.
<gene_1>:<effect_1>|... A gene can be repeated.
Effect details for each affected transcript. Format: < transcript 1 >:<gene 1>:<effect 1>:<details 1>|...
Annotator to identify the effect of the variant on protein coding.
Files
| Filename | Size | md5 |
|---|---|---|
| T2T_clinical_annotation.yaml | 2.81 KB | b71f6defc482c3c6bff21ebb81486b06 |
| genomic_resource.yaml | 248.0 B | a3c381b4b790a9cb114ca415f8e4d8c9 |
| statistics/ |