Resource
| Id | pipeline/hg38_demo_pipeline |
|---|---|
| Type | annotation_pipeline |
| Version | 0 |
| Summary |
Annotation Pipeline used in the paper.
|
| Description |
This is the example pipeline used in the GAIn paper. It includes a variety of annotators, such as effect annotation, position scoring, allele scoring, gene scoring, gene set annotation, and liftover annotation. The pipeline is designed to demonstrate the capabilities of the GAIn framework for genomic annotation. |
| Labels |
Pipeline Documentation
preamble
| Summary | Demo pipeline |
|---|---|
| Description | Demonstrates a GAIn pipeline |
| Input reference genome | hg38/genomes/ucsc-hg38 |
Annotators
Worst effect across all transcripts.
List of all genes
Annotator to identify the effect of the variant on protein coding.
The score is a number that reflects the conservation at a position.
position_aggregator: mean [default]
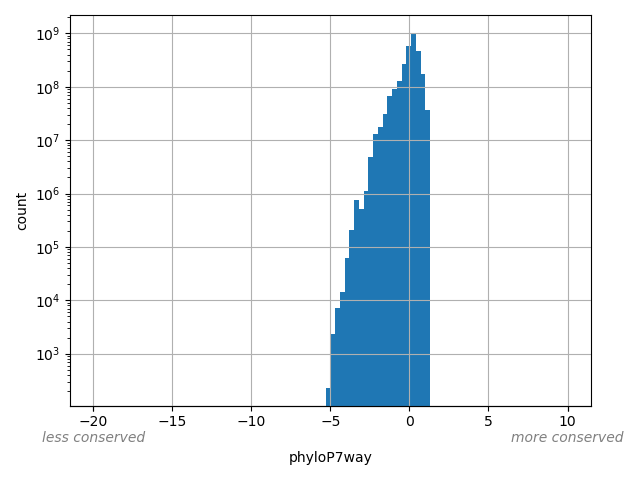
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
Normalized allele.
Alternate allele frequency
allele_aggregator: max
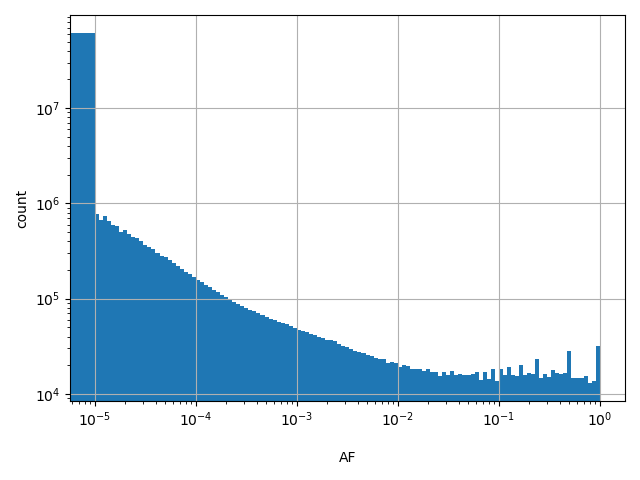
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
MVP scores were ranked among all MVP scores in dbNSFP. The rankscore is the ratio of the rank of the score over the total number of MVP scores in dbNSFP.
allele_aggregator: max
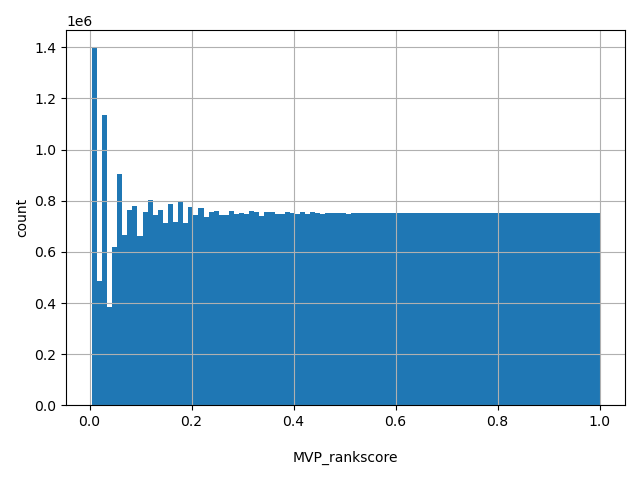
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
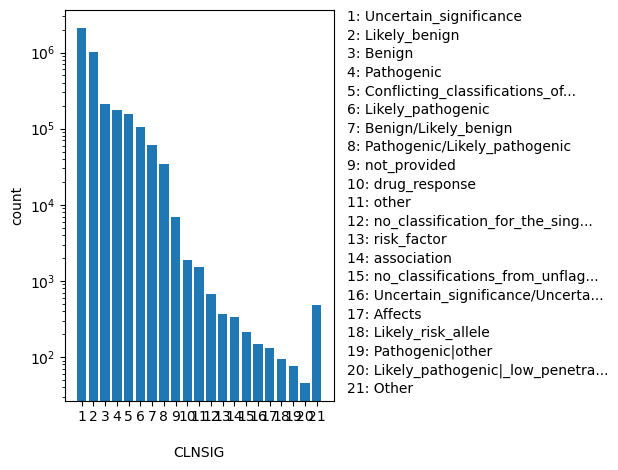
Aggregate germline classification for this single variant; multiple values are separated by a vertical bar
allele_aggregator: list
ClinVar's preferred disease name for the concept specified by disease identifiers in CLNDISDB
allele_aggregator: list
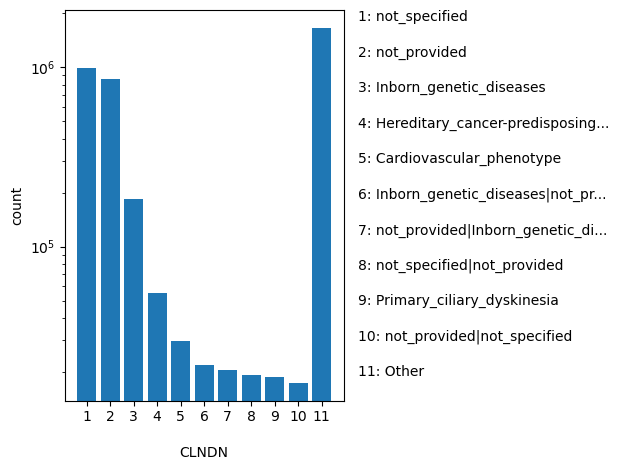
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
normalized_allele
Gene rank after sorting by pLI intolerance score
(87) http://www.gsea-msigdb.org/gsea/msigdb/cards/KEGG_APOPTOSIS
This gene set collection annotator uses the MSigDB_curated gene set collection.
The lifted over annotatable
Annotator to lift over a variant from one reference genome to another.
Missense badness, PolyPhen-2, and Constraint. A deleteriousness prediction score for missense variants"
allele_aggregator: max
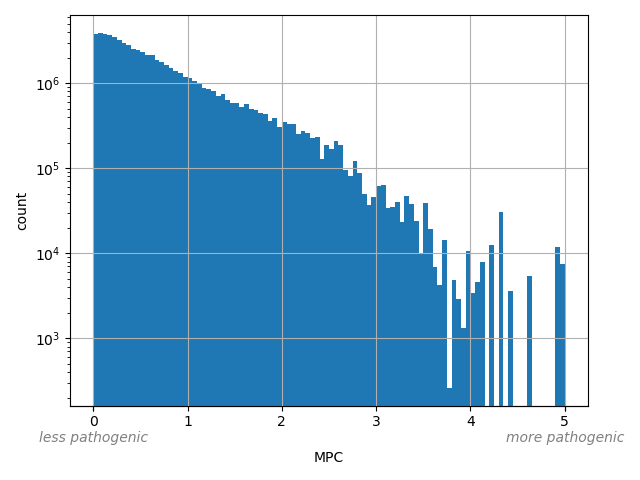
Annotator to use with scores that depend on allele like variant frequencies, etc.
Mode (mode parameter, applies to VCFAllele inputs only):
allele(default): exact chrom/pos/ref/alt match.region: aggregates scores for all allele lines overlapping the annotatable's span.
Non-VCFAllele annotatables always use region aggregation.
- input_annotatable:
hg19_annotatable
TFChIP-seq ENCSR000ATT [biosamplesummary="Homo sapiens K562" and target="CREBBP"]
position_aggregator: mean
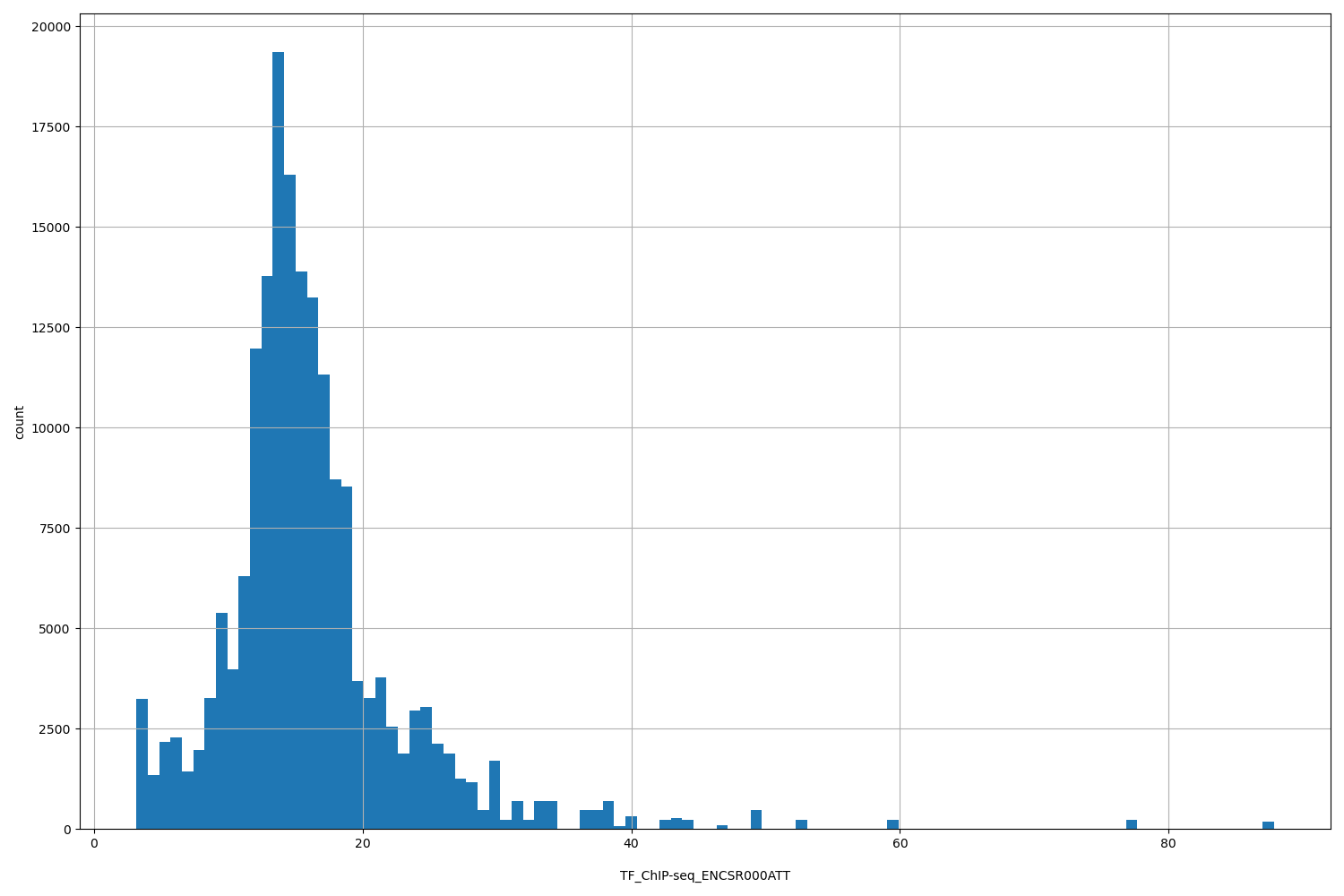
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR000BSN [biosamplesummary="Homo sapiens H1" and target="CREB1"]
position_aggregator: mean
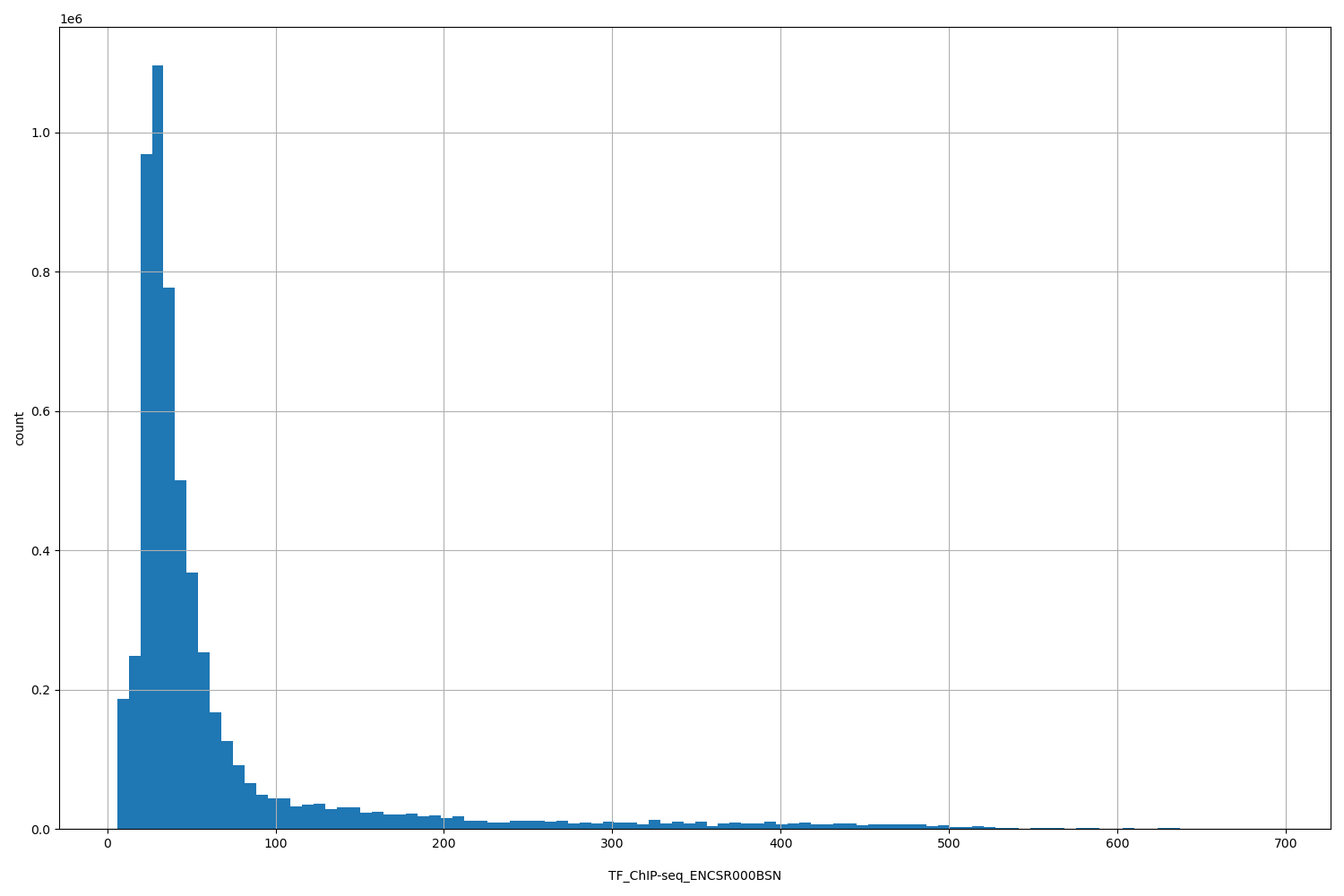
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR000BSO [biosamplesummary="Homo sapiens K562" and target="CREB1"]
position_aggregator: mean
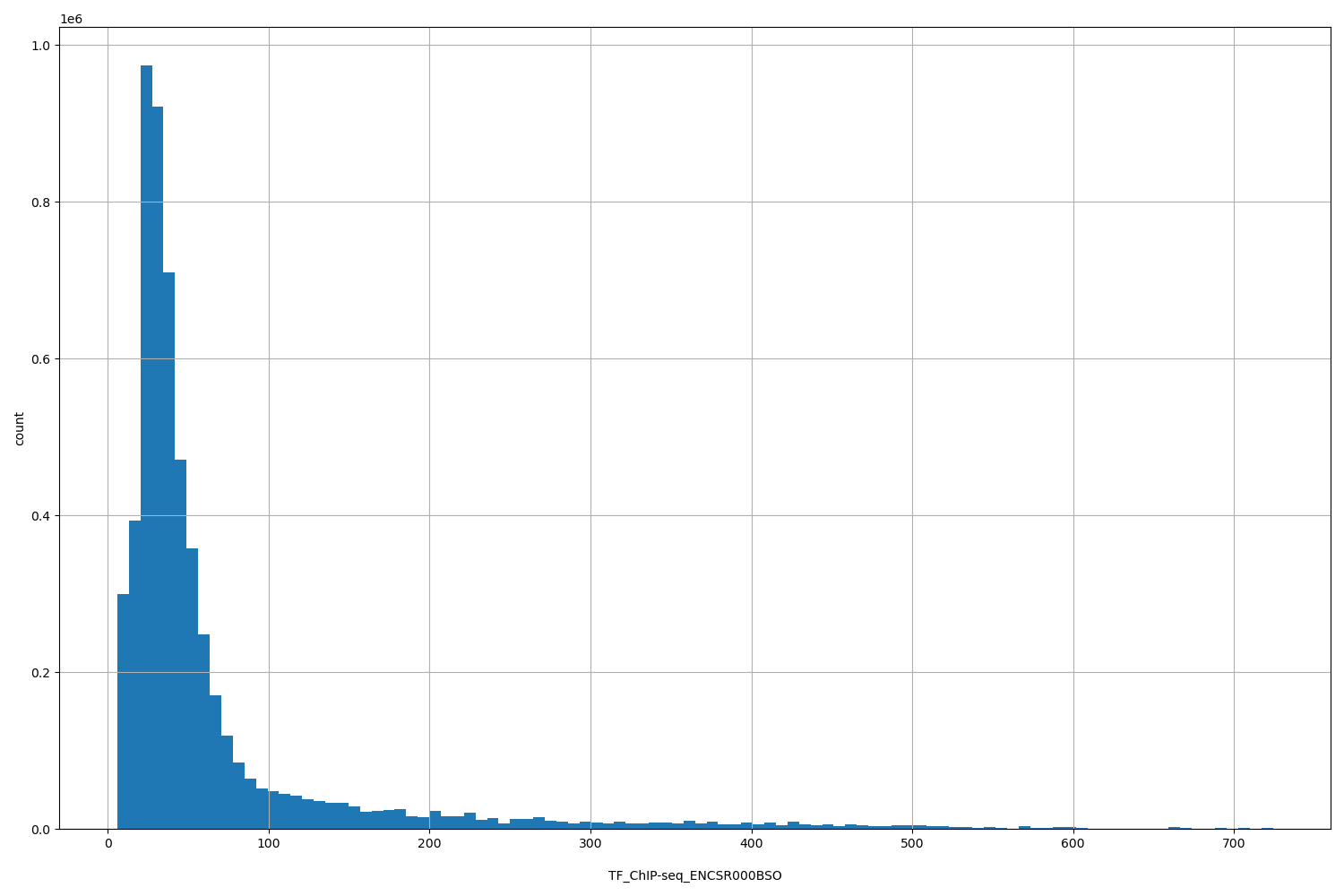
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR000BUF [biosamplesummary="Homo sapiens GM12878" and target="CREB1"]
position_aggregator: mean
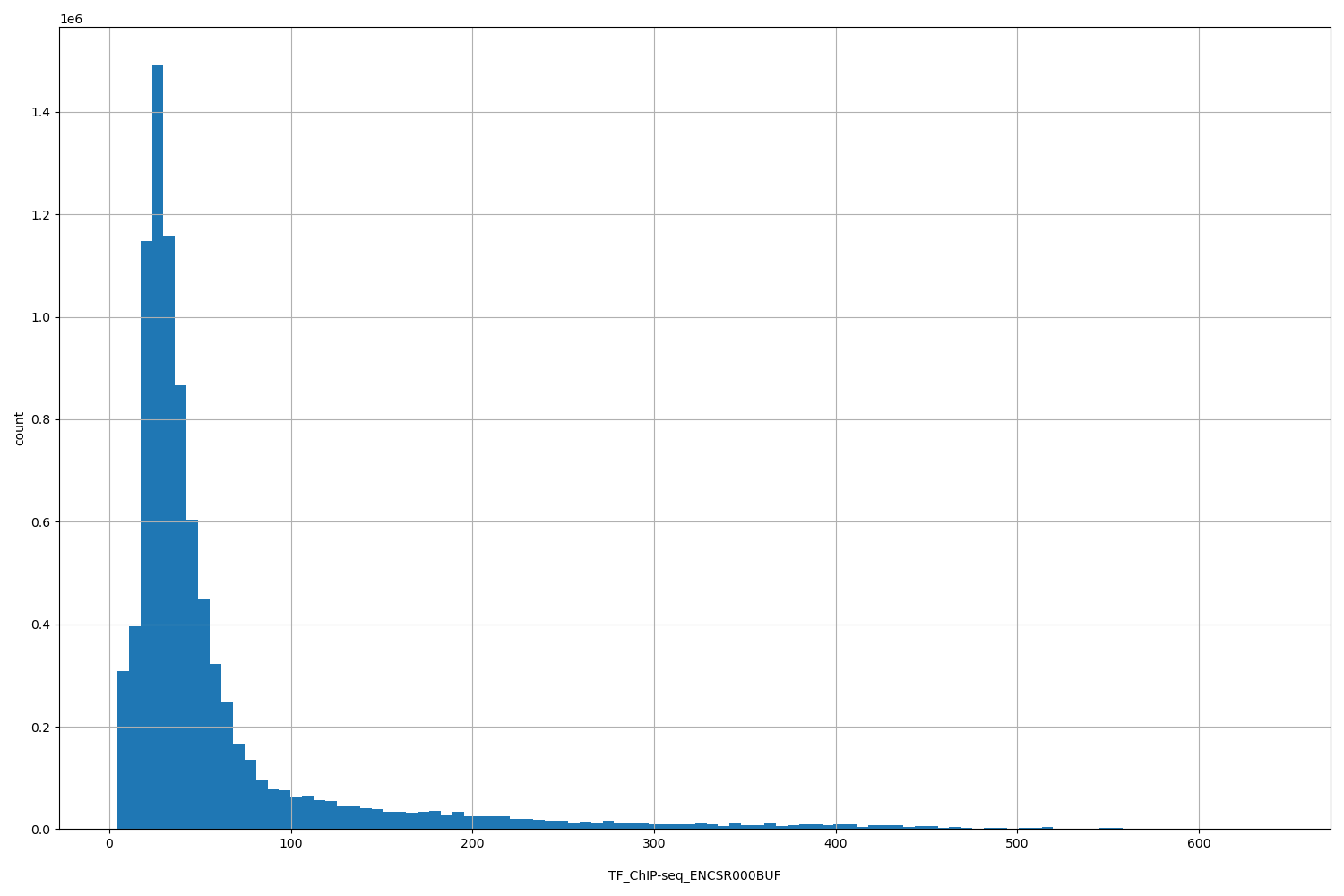
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR000BUR [biosamplesummary="Homo sapiens Ishikawa" and target="CREB1"]
position_aggregator: mean
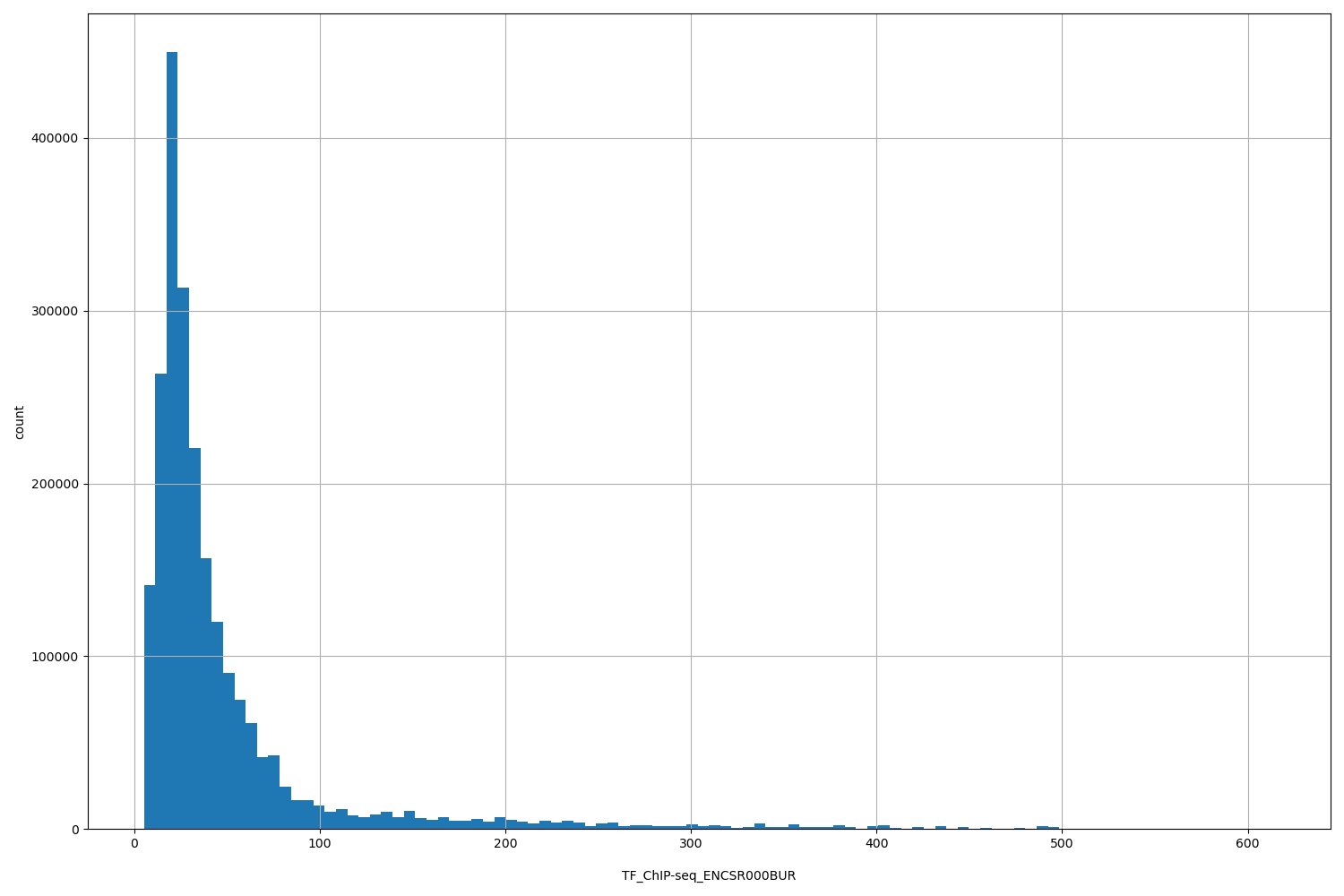
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR000BVL [biosamplesummary="Homo sapiens HepG2" and target="CREB1"]
position_aggregator: mean
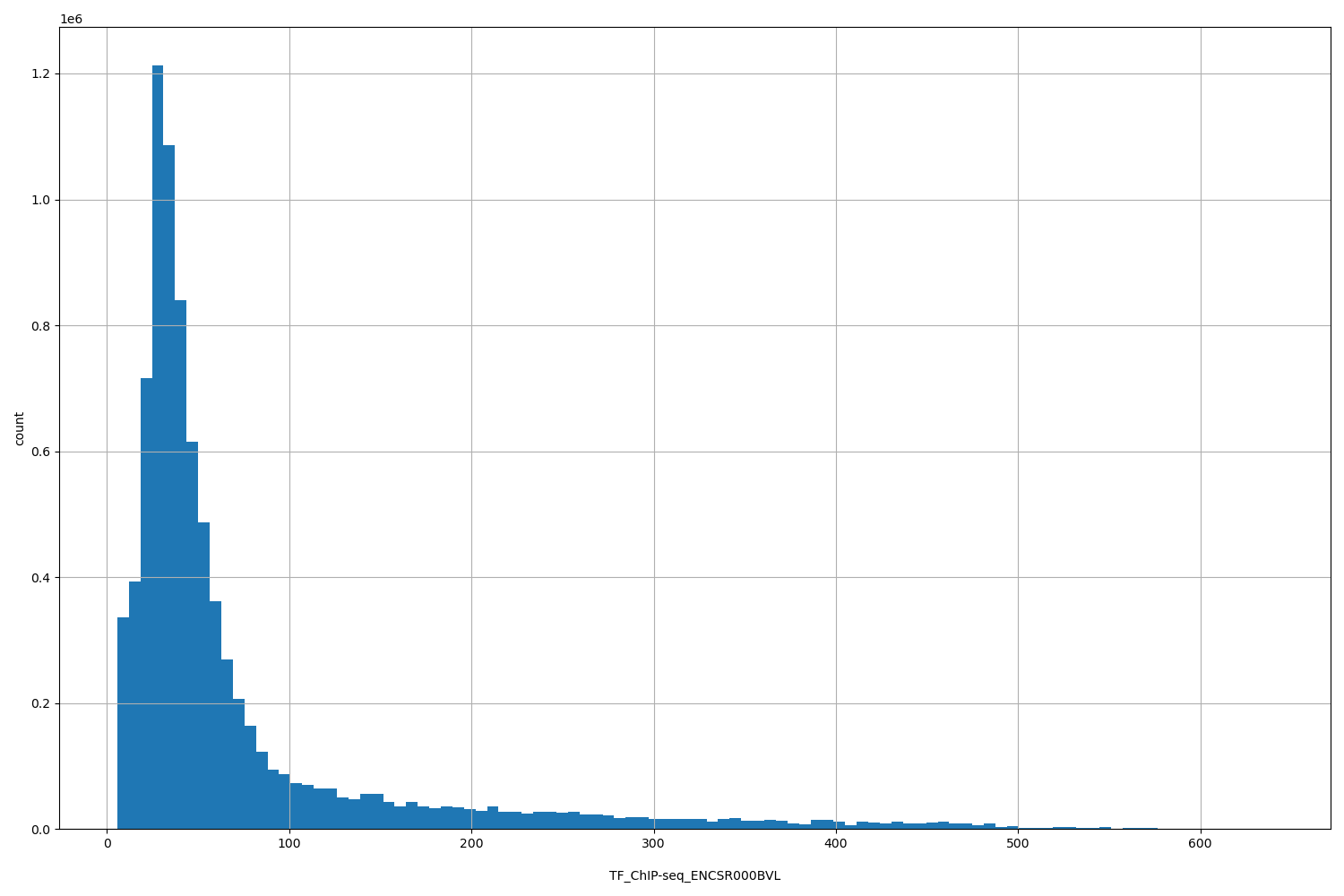
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR093FKD [biosamplesummary="Homo sapiens K562 stably expressing CREB3" and target="CREB3"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR109YGM [biosamplesummary="Homo sapiens K562" and target="CREB3L1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR112ALD [biosamplesummary="Homo sapiens HepG2" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR214ZAV [biosamplesummary="Homo sapiens GM23338 genetically modified (insertion) using CRISPR targeting H. sapiens CREB1" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR237OGW [biosamplesummary="Homo sapiens HepG2 genetically modified (insertion) using CRISPR targeting H. sapiens CREBL2" and target="CREBL2"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR331ORD [biosamplesummary="Homo sapiens HepG2 genetically modified (insertion) using CRISPR targeting H. sapiens CREB1" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR471WXT [biosamplesummary="Homo sapiens K562 genetically modified (insertion) using CRISPR targeting H. sapiens CREB1" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR620DUQ [biosamplesummary="Homo sapiens MCF-7" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR700LFA [biosamplesummary="Homo sapiens WTC11 genetically modified (insertion) using CRISPR targeting H. sapiens CREB1" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR758GOA [biosamplesummary="Homo sapiens SK-N-SH genetically modified\ \ (insertion) using CRISPR targeting H. sapiens CREB5 treated with 6 \u03BCM all-trans-retinoic\ \ acid for 48 hours" and target="CREB5"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR855XFL [biosamplesummary="Homo sapiens HepG2 genetically modified (insertion) using CRISPR targeting H. sapiens CREB3" and target="CREB3"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR897JAS [biosamplesummary="Homo sapiens MCF-7" and target="CREB1"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
TFChIP-seq ENCSR935PEA [biosamplesummary="Homo sapiens K562 genetically modified (insertion) using CRISPR targeting H. sapiens CREB5" and target="CREB5"]
position_aggregator: mean
Annotator to use with genomic scores depending on genomic position like phastCons, phyloP, FitCons2, etc.
Files
| Filename | Size | md5 |
|---|---|---|
| genomic_resource.yaml | 486.0 B | 2485f54375717b4a00a7c18f498b7cef |
| hg38_demo_pipeline.yaml | 1.43 KB | 4b42ea404977eedf8d92ca747aa26222 |
| statistics/ |